About
A data analytics specialist with over 5 years of experience in genetics and genomics, statistical genetics, applied biostatistics and statistical method development. Expertise in molecular and statistical genetics, with a focus on development of methods improving the utility and equity of genomic resources. Passionate about leveraging data-driven approaches to advance healthcare and improve patient lives globally.
Areas of Expertise
Data analytics and software development
R, Python, Java, SQLHuman and molecular genetics
Variant analysis, NGS technologies and interpretation, common complex diseases and traitsVersion control and collaboration
Git, Github, BioconductorOral, written and verbal communication
Microsoft office, GSuite, manuscript writing, oral presentationProject management
Time management, communication, organization, task prioritization
Professional Experience
Bioinformatics Research Professional
2025-present
University of Colorado - Denver | Anschutz Medical Campus
Human Medical Genetics and Genomics ProgramResearcher and peer mentor for biostatistics and statistical genetics, developing statistical methods to improve equity and re-use of summary genetic data.
Developed R software and statistical test to estimate and evaluate local substructure proportions from allele frequency data. Tested in extensive simulations and real data applications. Wrote co-first authored peer reviewed manuscript to share software and results.
Developed R software and methods to reconstruct case and control allele frequencies from GWAS summary statistics.
Created an R software package made publicly available through Github and Bioconductor.
Wrote first author manuscript to share software and results (under review).
Mentored research assistants, undergraduates, and junior graduate students to promote development of skills in genetics, statistical data analysis, and independent research.
Research Intern
2018-2020
Illumina IncPerforming molecular genetics, genetic engineering, and deep phenotyping in model organisms.
Large scale genomic sequencing and phenotyping of over 1000 strains of S. cerevisiae wild isolates, results published in Science.
Utilization and experimental validation of CRISPR-Cas9 bioinformatics tool, resulting in first author peer-reviewed manuscript
Undergraduate Researcher
2017-2019
University of Wisconsin - Madison, Department of Horticulture; USDA-ARSUndergraduate researcher studying genetics of carrots
Contributed to experimental setup and data collection for studies of draught resistance in carrot
Performed laboratory experiments including HPLC, DNA and RNA extraction, DNA library preparation, and cloning
Completed mentored research study of genotype data to determine QTL for nematode resistance in carrots
Education
University of Colorado - Denver | Anschutz Medical Campus
2020 - 2024
Ph.D. - Human Medical Genetics and Genomics Program
Dissertation: "Advancing methodologies for estimating substructure and allelel frequencies from genetic summary data"
Thesis Advisor: Audrey E Hendricks, Ph.D.
University of Wisconsin - Madison
2016 - 2019
Major: Genetics and GenomicsMinors: Computer Science, Integrated Studies of Science Engineering and Society
External Presentations
First author abstracts presented externally of the university

ASHG 2024
Denver, CO, USA
Poster presentation
WNAR 2024
Fort Collins, CO, USA
Oral presentation; student paper competition
JSM 2023 Toronto, Canada
Oral presentation
ASHG 2023
Washington DC, USA
Poster presentation
IGES 2022
Paris, France
Poster presentation
Publications
"Characterizing substructure via mixture modeling in large-scale genetic summary statistics"
2025 - American Journal of Human GeneticsRead
"CCAFE: Estimating Case and Control Allele Frequencies from GWAS Summary Statistics"
2024 - bioRXivRead
"Genomic factors shape carbon and nitrogen metabolic niche breadth across Saccharomycotina yeasts"
2024 - ScienceRead
"CRISpy-Pop: A Web Tool for Designing CRISPR/Cas9-Driven Genetic Modifications in Diverse Populations"
2020 - G3: Genes | Genomes | GeneticsRead
Professional Societies
American Society of Human GeneticsAmerican Statistical AssociationInternational Genetic Epidemiology SocietyWomen's Association of Computing Machinery

Projects
Here you’ll find a selection of my work. Explore my projects to learn more about what I do, and check out the links of full project reports, papers, and code.
Summix2
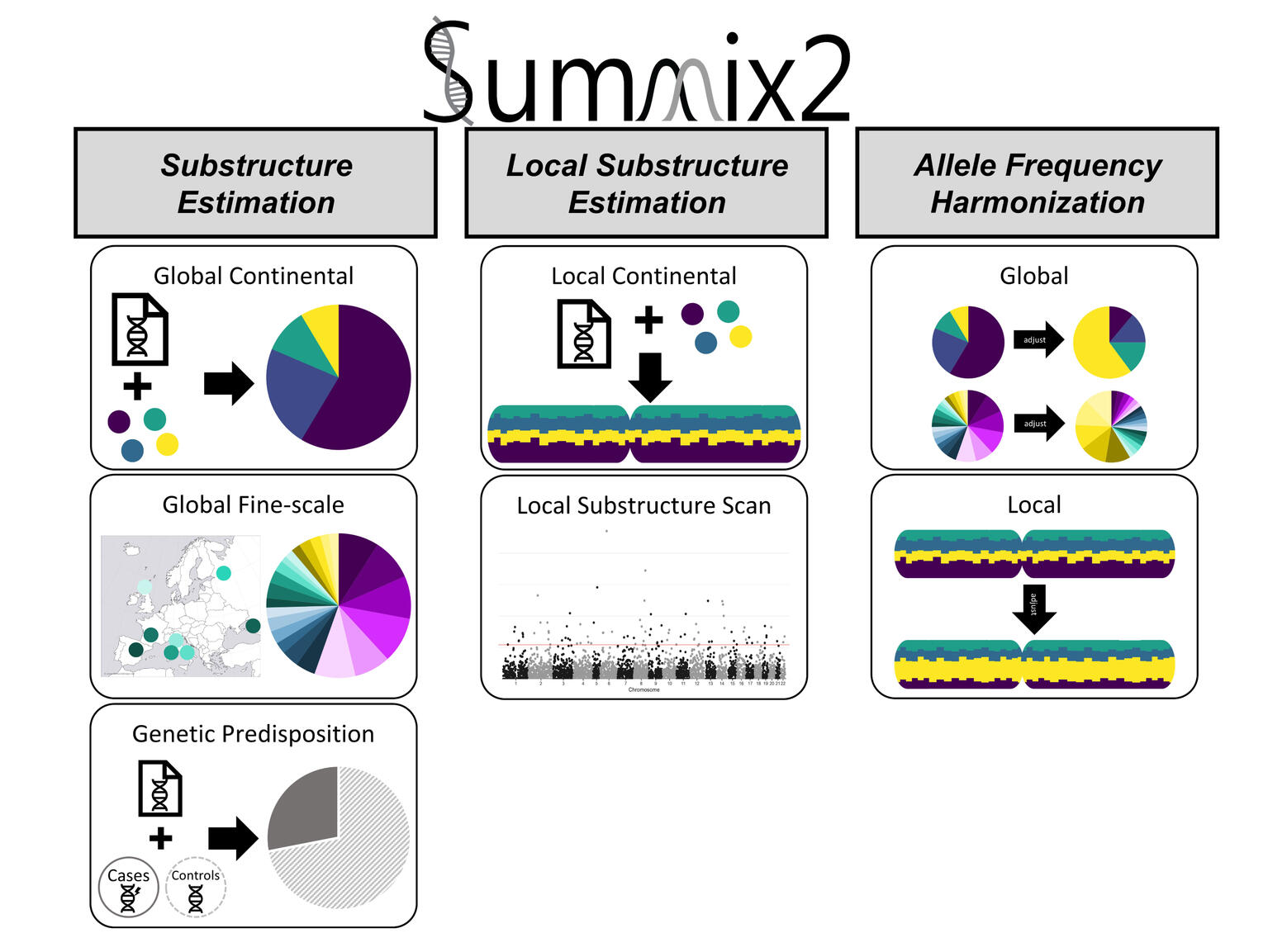
A comprehensive set of methods and software based on a computationally efficient mixture model to enable the harmonization of genetic summary data by estimating and adjusting for substructure.
CCAFE
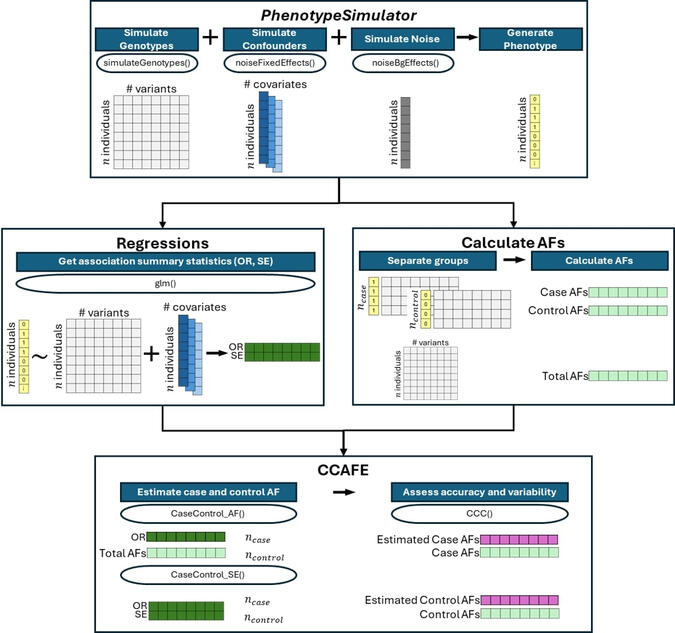
Two frameworks to derive case and control AFs from GWAS summary statistics using the odds ratio, case and control sample sizes, and either the total (case and control aggregated) AF or standard error (SE).
Case Case GWAS
(CC-GWAS)

Genetic summary statistics, such as case and control allele frequencies (AFs) can be used for secondary analyses like case-case genome wide association studies (CC-GWAS).I have adapted their code and framework from C and implemented it in R.
Regression Confounding Correction
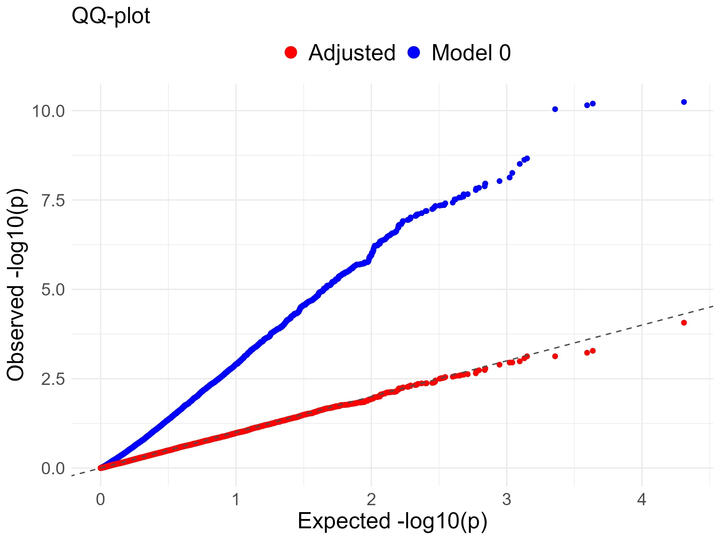
Residual substructure can lead to confounding in GWAS.
I have implemented this framework and tested it in various new settings representing continental-level substructure in simulations.
Diabetes Risk Substructure
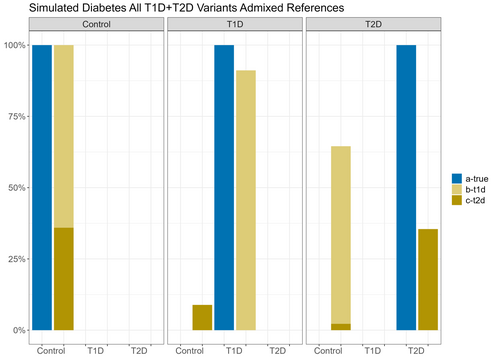
In order to assess the ability to estimate disease risk substructure, we performed simulations for Type 1 Diabetes (T1D) and Type 2 Diabetes (T2D).
Simulations for Local Ancestry Estimation
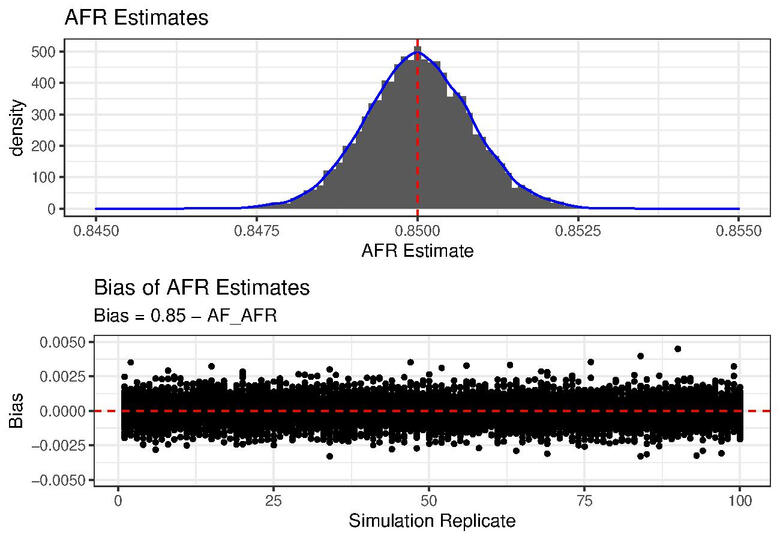
For this study I used the data in the Mimic3 demo electronic health record (EHR) dataset define the outcome variable as death during the ICU stay.
EHR - Predicting death during ICU Stay
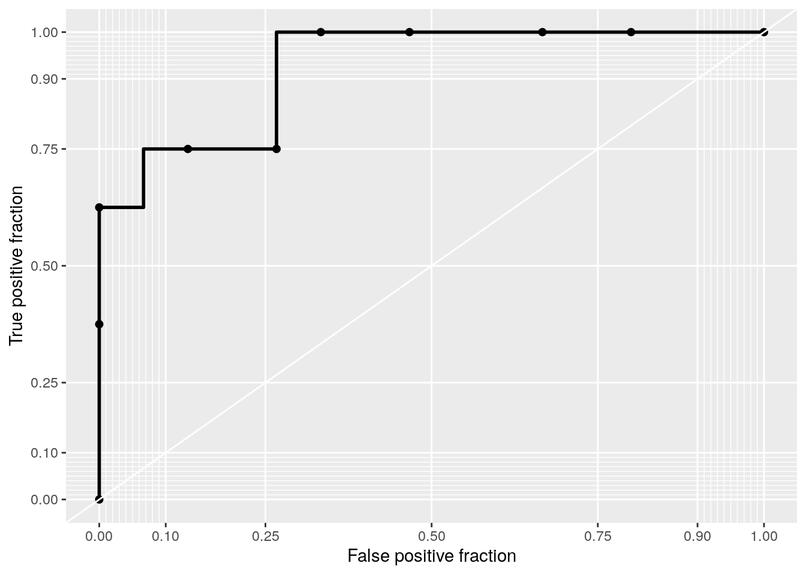
For this study I used the data in the Mimic3 demo electronic health record (EHR) dataset define the outcome variable as death during the ICU stay.
EHR - Diabetic Complications
For this study I used corpus of EHR clinical text notes identify patients with diabetic complications.
ML Prediction Models IPF

This study aimed to identify gene expression and DNA methylation biomarkers in whole blood that could predict risk for idiopathic pulmonary fibrosis (IPF) before irreversible lung damage occurs.
scRNA-seq Analysis
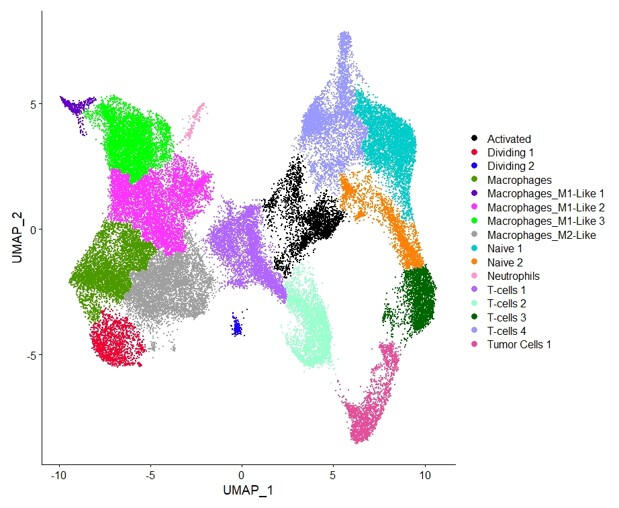
This study explores host intrinsic differences in immune response that influence the effectiveness of anti-PD-L1 treatment in head and neck squamous cell carcinoma (HNSCC).
Project Type
Ph.D. Thesis Research
Date
February 2021 - January 2025
Location
University of Colorado - Denver,
Anschutz Medical Campus
Role
Co-first author
Summix2
Genetic summary data are broadly accessible and highly useful, including for risk prediction, causal inference, fine mapping, and incorporation of external controls. However, collapsing individual-level data into summary data, such as allele frequencies, masks intra- and inter-sample heterogeneity, leading to confounding, reduced power, and bias. Ultimately, unaccounted-for substructure limits summary data usability, especially for understudied or admixed populations. There is a need for methods to enable the harmonization of summary data where the underlying substructure is matched between datasets. Here, we present Summix2, a comprehensive set of methods and software based on a computationally efficient mixture model to enable the harmonization of genetic summary data by estimating and adjusting for substructure. In extensive simulations and application to public data, we show that Summix2 characterizes finer-scale population structure, identifies ascertainment bias, and scans for potential regions of selection due to local substructure deviation. Summix2 increases the robust use of diverse, publicly available summary data, resulting in improved and more equitable research.

CCAFE
Methods involving summary statistics in genetics can be quite powerful but can be limited in utility. For instance, many post-hoc analyses of disease studies require case and control allele frequencies (AFs), which are not always published. We present two frameworks to derive case and control AFs from GWAS summary statistics using the odds ratio, case and control sample sizes, and either the total (case and control aggregated) AF or standard error (SE). In simulations and real data, derivations of case and controls AFs using total AF is highly accurate across all settings (e.g., minor AF, condition prevalence). Conversely, derivations using SE underestimate common variant AFs (e.g. minor allele frequency >0.3) in the presence of covariates. We develop an adjustment using gnomAD AFs as a proxy for true AFs, which reduces the bias when using SE. While estimating case and control AFs using the total AF is preferred due to its high accuracy, estimating from the SE can be used more broadly since SE can be derived from p-values and beta estimates, which are commonly provided. The methods provided here expand the utility of publicly available genetic summary statistics and promote the reusability of genomic data. The R package CCAFE, with implementations of both methods, is freely available on Bioconductor and GitHub.

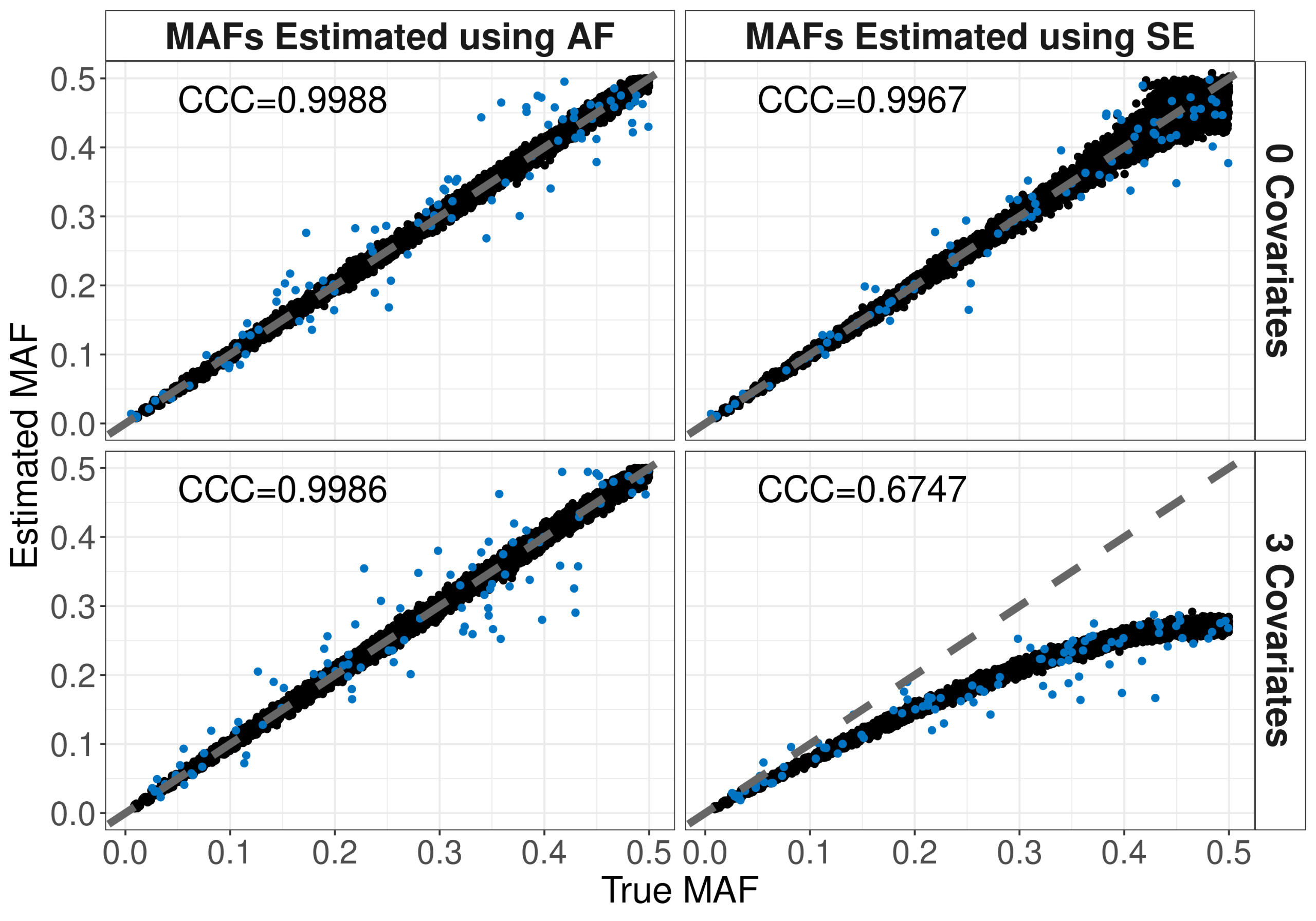
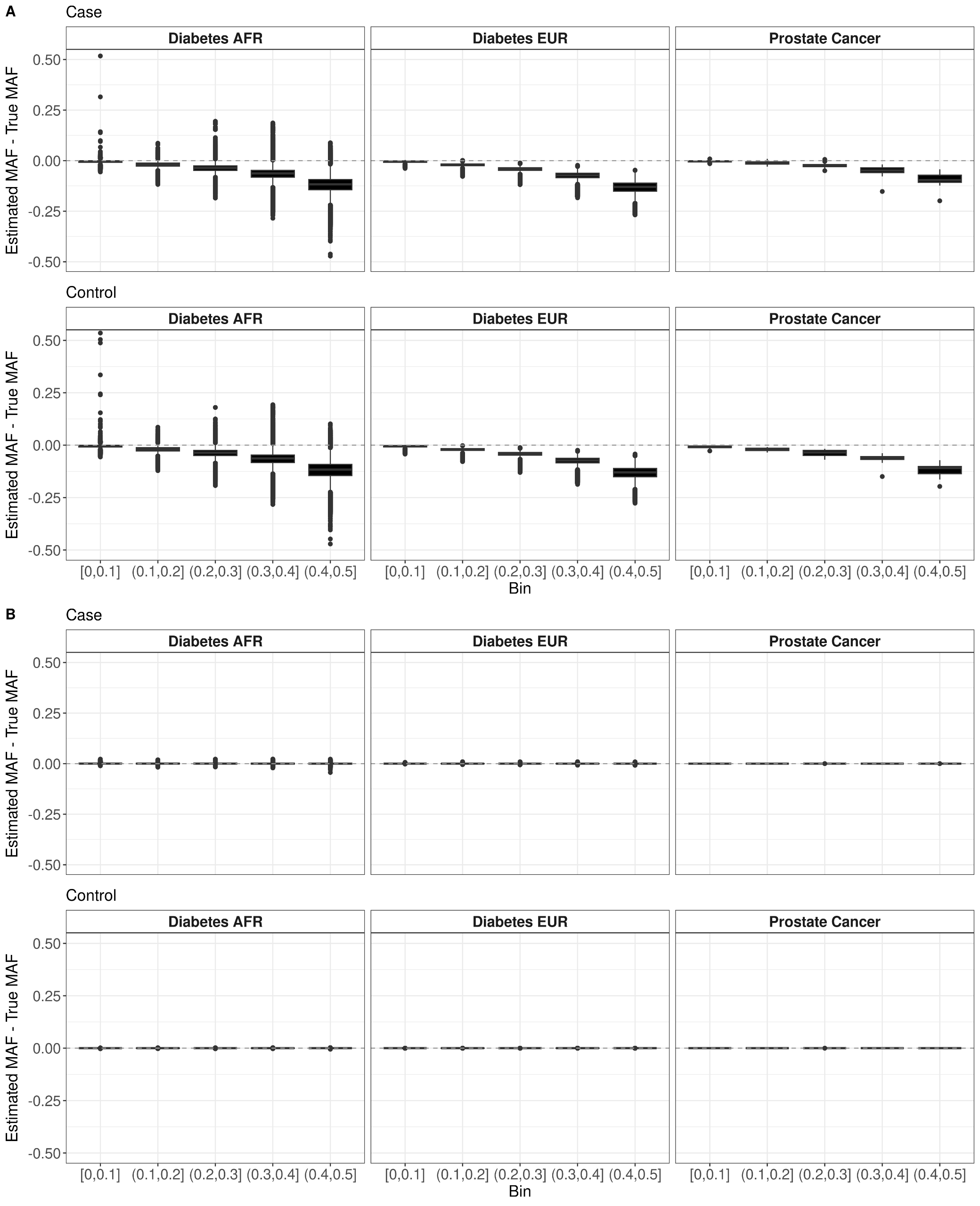
Project Type
Genomic summary data analysis; coding
Date
February 2024 - October 2024
Location
University of Colorado - Denver,
Anschutz Medical Campus
Role
Project Lead
Link
Case Case GWAS (CC-GWAS)
Genetic summary statistics, such as case and control allele frequencies (AFs) can be used for secondary analyses like case-case genome wide association studies (CC-GWAS). CC-GWAS identifies genetic variants that have different AFs between two samples of cases with related (but different) traits or diseases. This could provide insight as to why a person might develop one trait instead of a closely related second trait. CC-GWAS was first introduced by Peyrot and Price in 2021. A second alternative framework to perform CC-GWAS was proposed by Yang et al. in 2022. Yang et al provided C-based software to perform this analysis as part of a larger software package called ReACt. I have adapted their code and framework from C and implemented it in R. This project demonstrates to utility of the CCAFE software to improve data reuse.
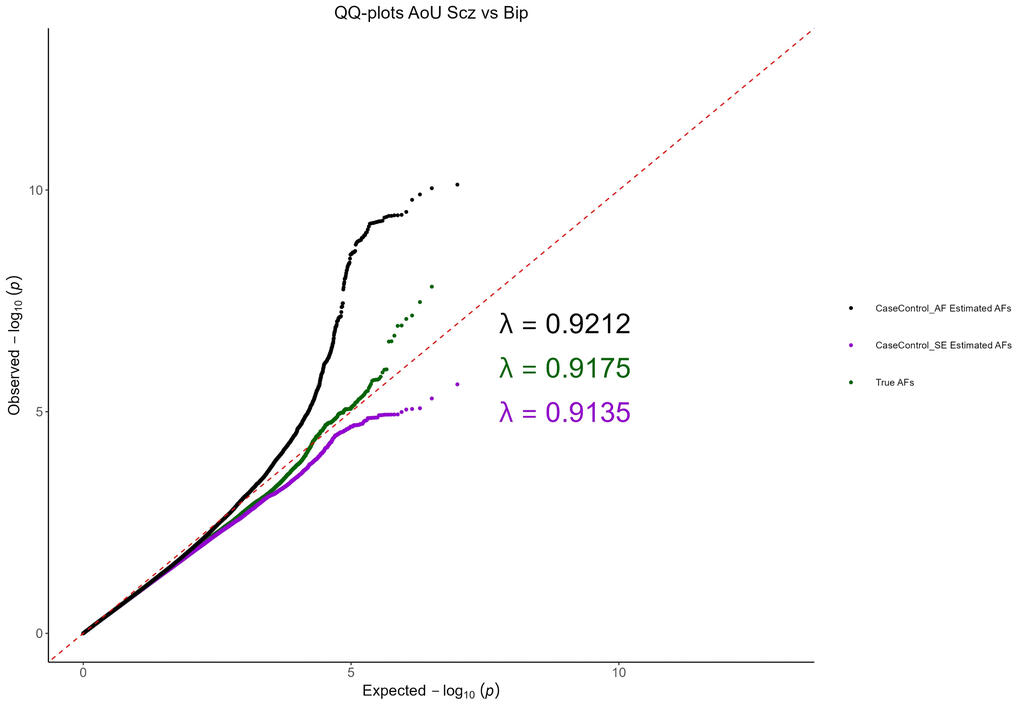
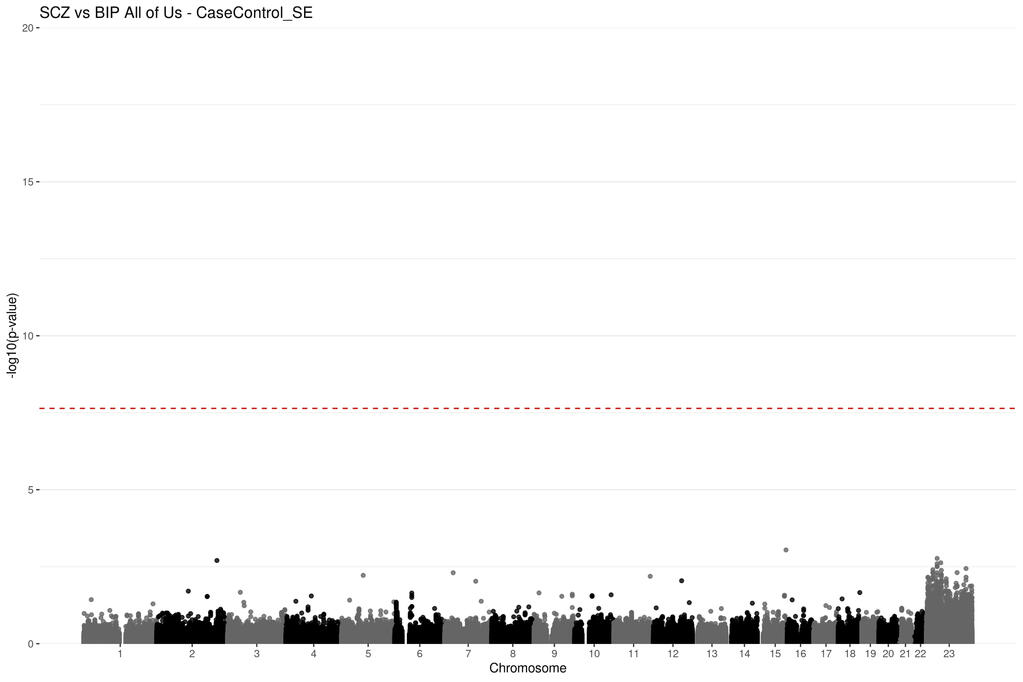
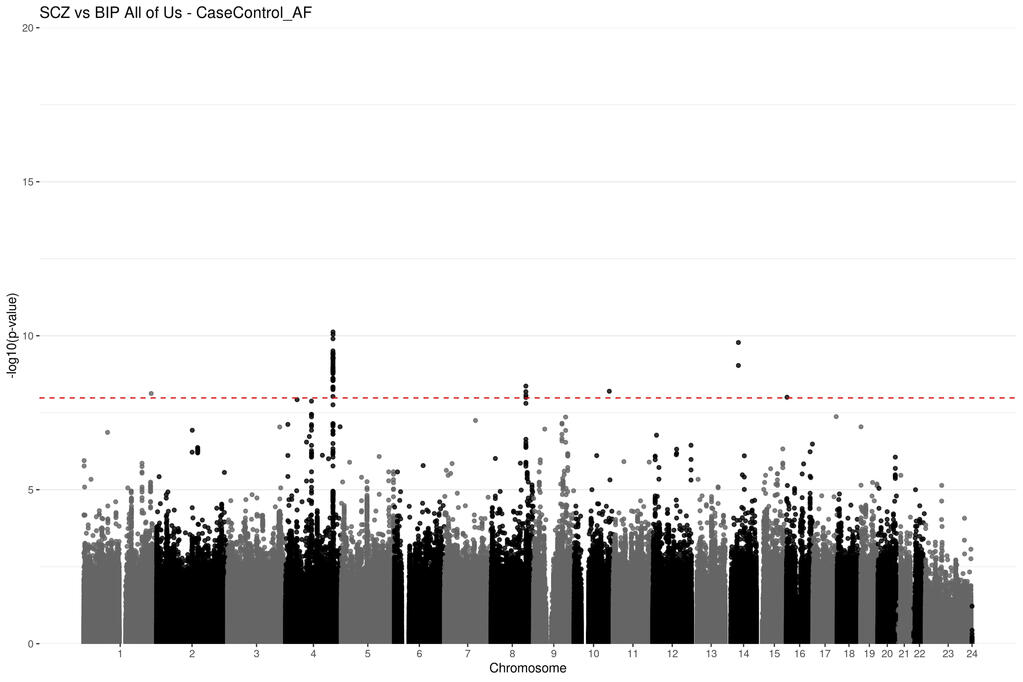
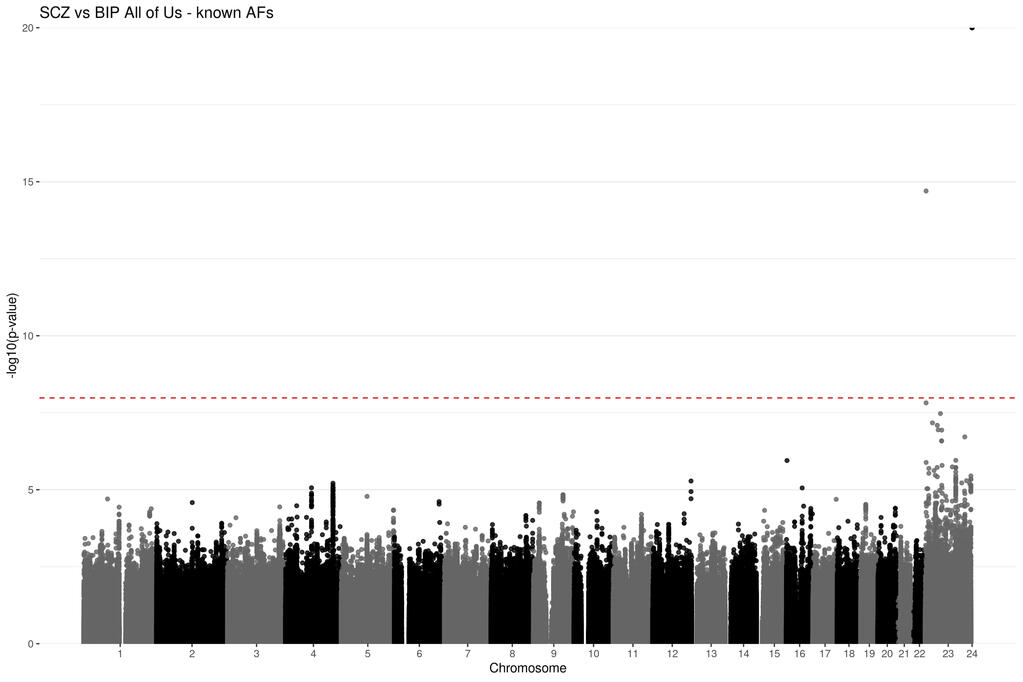
Project Type
Regression; Simulation
Date
September 2023 - February 2024
Location
University of Colorado - Denver,
Anschutz Medical Campus
Link
Regression Confounding Correction
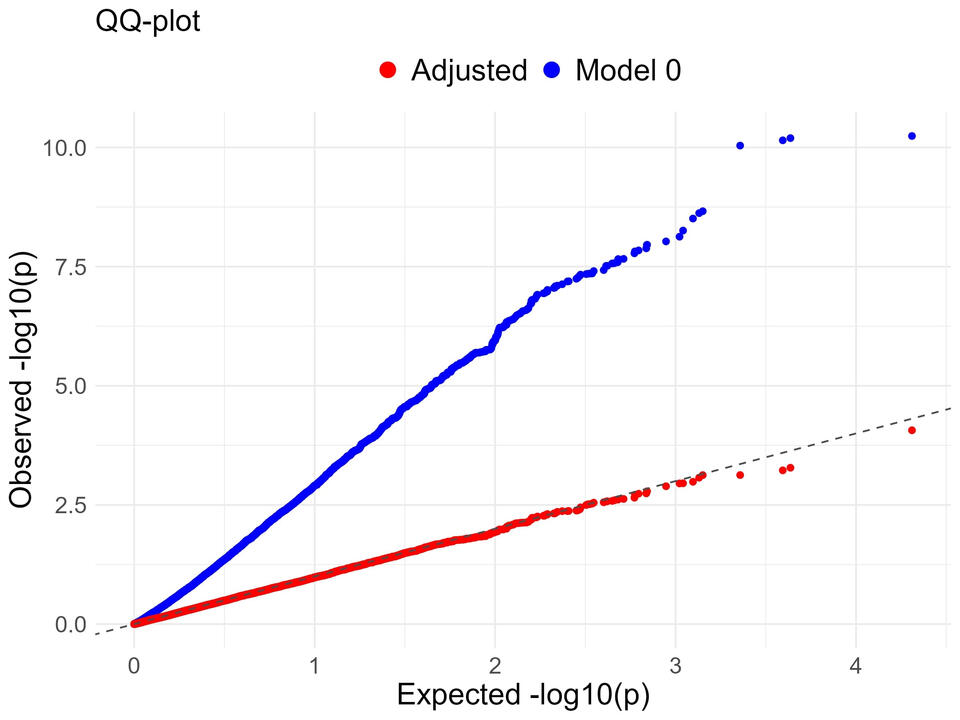
Residual substructure can lead to confounding in GWAS. In 2022, Yung derived a direct adjustment which can derive new beta estimations that account for population structure. I have implemented this framework and tested it in various new settings representing continental-level substructure in simulations.
Project Type
Summix2 application; Genetic summary data analysis; Simulations
Date
August 2023 - February 2024
Location
University of Colorado - Denver,
Anschutz Medical Campus
Role
Project Lead
Diabetes Risk Substructure
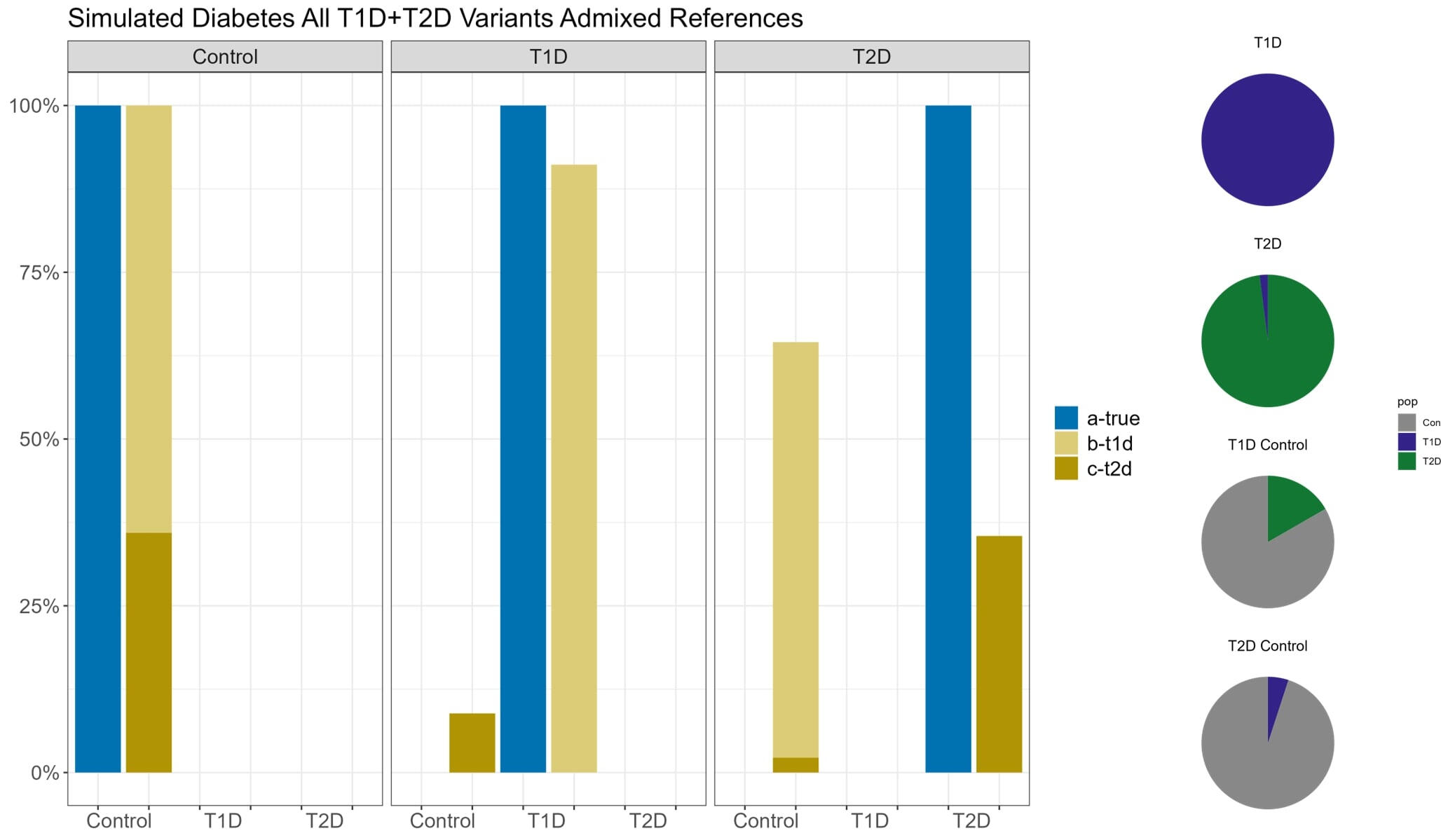
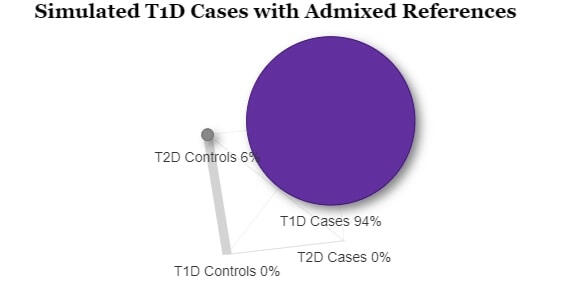
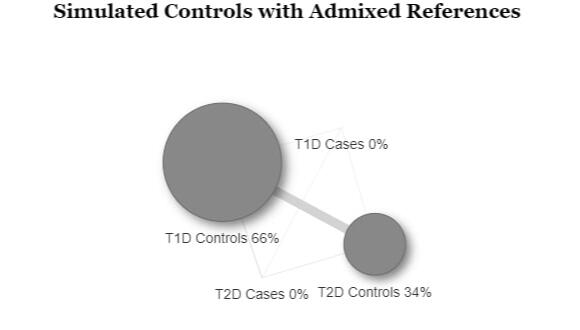
In order to assess the ability to estimate disease risk substructure, we performed simulations for Type 1 Diabetes (T1D) and Type 2 Diabetes (T2D). Using the Colorado Center for Personalized Medicine (CCPM) allele frequency (AF) data for individuals of European genetic ancestry, we simulated observed and reference samples. From CCPM we have AF data from individuals subset by age (18-30, 31-50, >50 years) and disease status (T1D, T2D, no T1D nor T2D). For cases, we used the AF data for all ages, and for controls we used the AF data only for individuals > 50 years to minimize the chances of including affected individuals that have yet to be diagnosed. Genotypes were simulated for each single nucleotide polymorphism (SNP) using rmultinom in R, using the CCPM data to calculate the probabilities assuming Hardy-Weinberg equilibrium.It is likely that existing datasets have misclassification within their case and control samples. To replicate this situation, we simulated reference samples that were a mixture of T1D cases, T2D cases, and controls. The T1D case reference sample was 100% T1D cases. The T2D case reference was 98% T2D cases and 5% T1D cases. The T1D control reference was 80% controls and 20% T2D cases, and the T2D controls were 95% controls and 5% T1D cases. We simulated observed samples that were 100% controls, T1D cases, and T2D cases. We then used Summix2 to estimate the genetic substructure proportions of these observed samples using the simulated mixed references. We find that we capture the network of similarity between the reference samples when we estimate the substructure, which can be represented as a network, where the edge weights are reciprocal of the pairwise Fst.
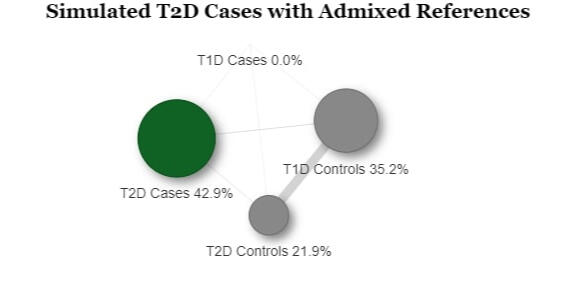
Project Type
Biostatistics; Simulation; Local Ancestry; Genetic summary data analysis
Date
December 2021
Location
University of Colorado - Denver,
Anschutz Medical Campus
Link
Simulations for Local Ancestry Estimation
This is where the project description goes. Give an overview or go in depth - what it's all about, what inspired you, how you created it, or anything else you'd like visitors to know. To add Project descriptions, go to Manage Projects.
Project Type
Electronic health records; Electronic medical records; Computational phenotyping; Clinical prediction; NLP; Regular expressions
Date
May 2021
Location
University of Colorado - Denver,
Anschutz Medical Campus
EHR - Predicting death during ICU stay
Using the data in the Mimic3 demo electronic health record (EHR) dataset define the outcome variable as death during the ICU stay. Create an algorithm to define the population. Then clinical predictors to predict the outcome and evaluate and validate the methodology and algorithms created.
Project Type
Electronic health records; electronic medical records; Regular expressions; text processing; NLP
Date
April 2021
Location
University of Colorado - Denver,
Anschutz Medical Campus
EHR - Diabetic complications
Using corpus of EHR clinical text notes identify patients with diabetic complications. To accomplish this task I determine an approach, write regular expressions, asses the test processing algorithm, and reflect on the results.
Project Type
AI and ML; Prediction models; Methylation; RNA sequencing
Date
February 2021
Location
University of Colorado - Denver,
Anschutz Medical Campus
Link
ML Prediction Models IPF
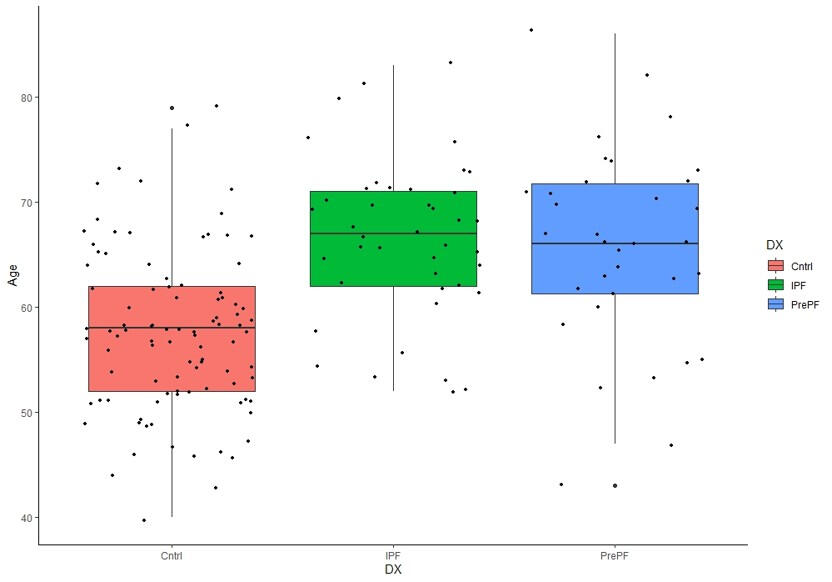
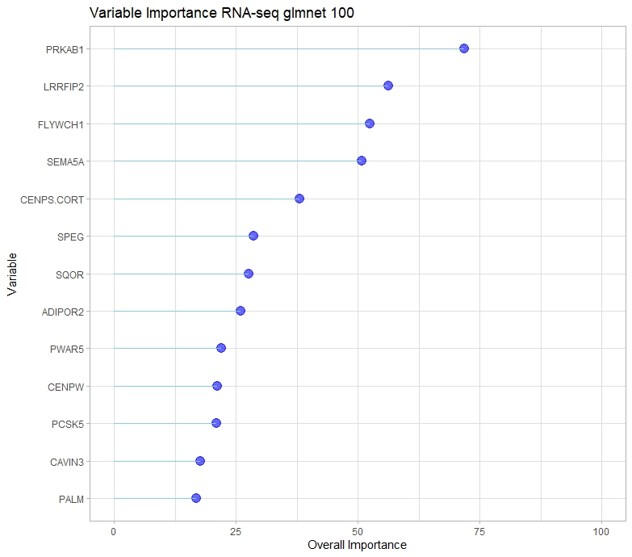
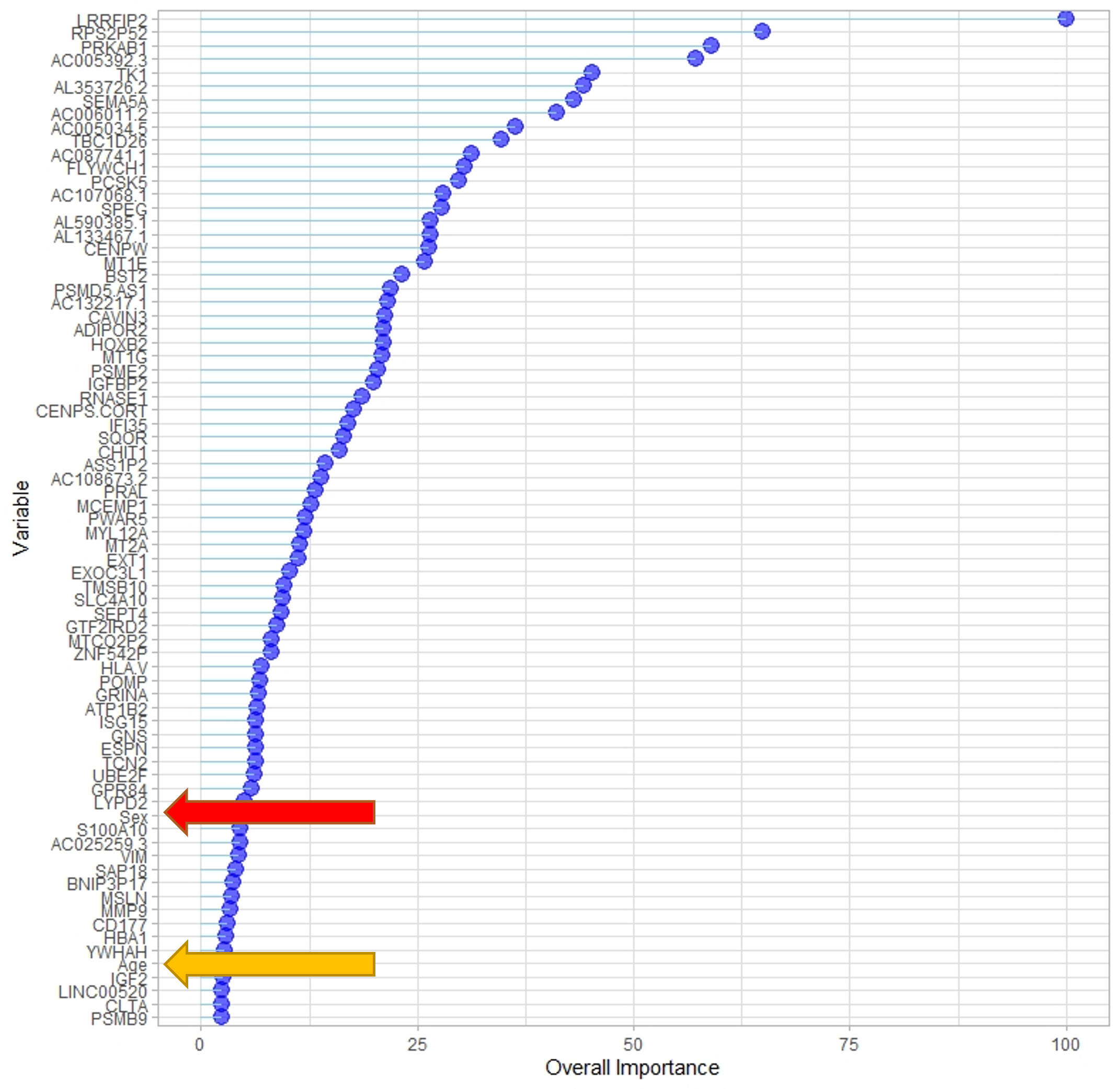
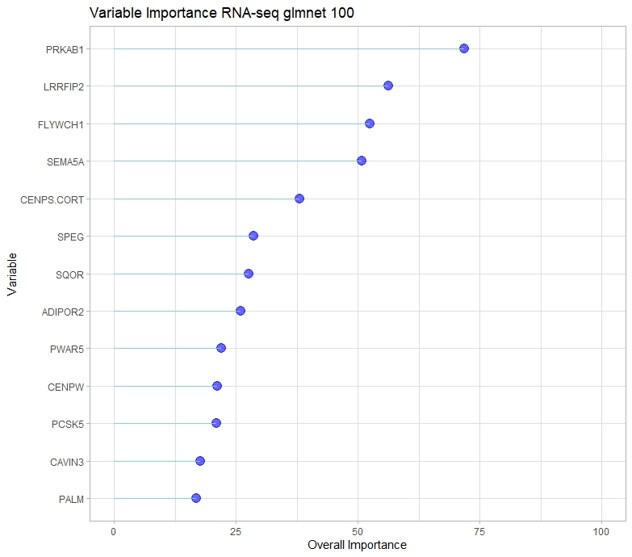
This study aimed to identify gene expression and DNA methylation biomarkers in whole blood that could predict risk for idiopathic pulmonary fibrosis (IPF) before irreversible lung damage occurs. Using RNA sequencing and DNA methylation data from the CADET cohort, machine learning models were trained to distinguish individuals with preclinical pulmonary fibrosis (PrePF) from controls. Differential expression and methylation analyses identified key genes and CpG sites associated with IPF risk, including SPEG, PCSK5, and MUC5B. The best predictive models achieved high accuracy and were robust to demographic confounders such as age and sex. These findings suggest that blood-based biomarkers could facilitate earlier IPF detection, potentially improving patient prognosis and guiding early intervention strategies.
Project Type
Rotation project; single cell RNA sequencing analysis
Date
November 2020
Location
University of Colorado - Denver,
Anschutz Medical Campus
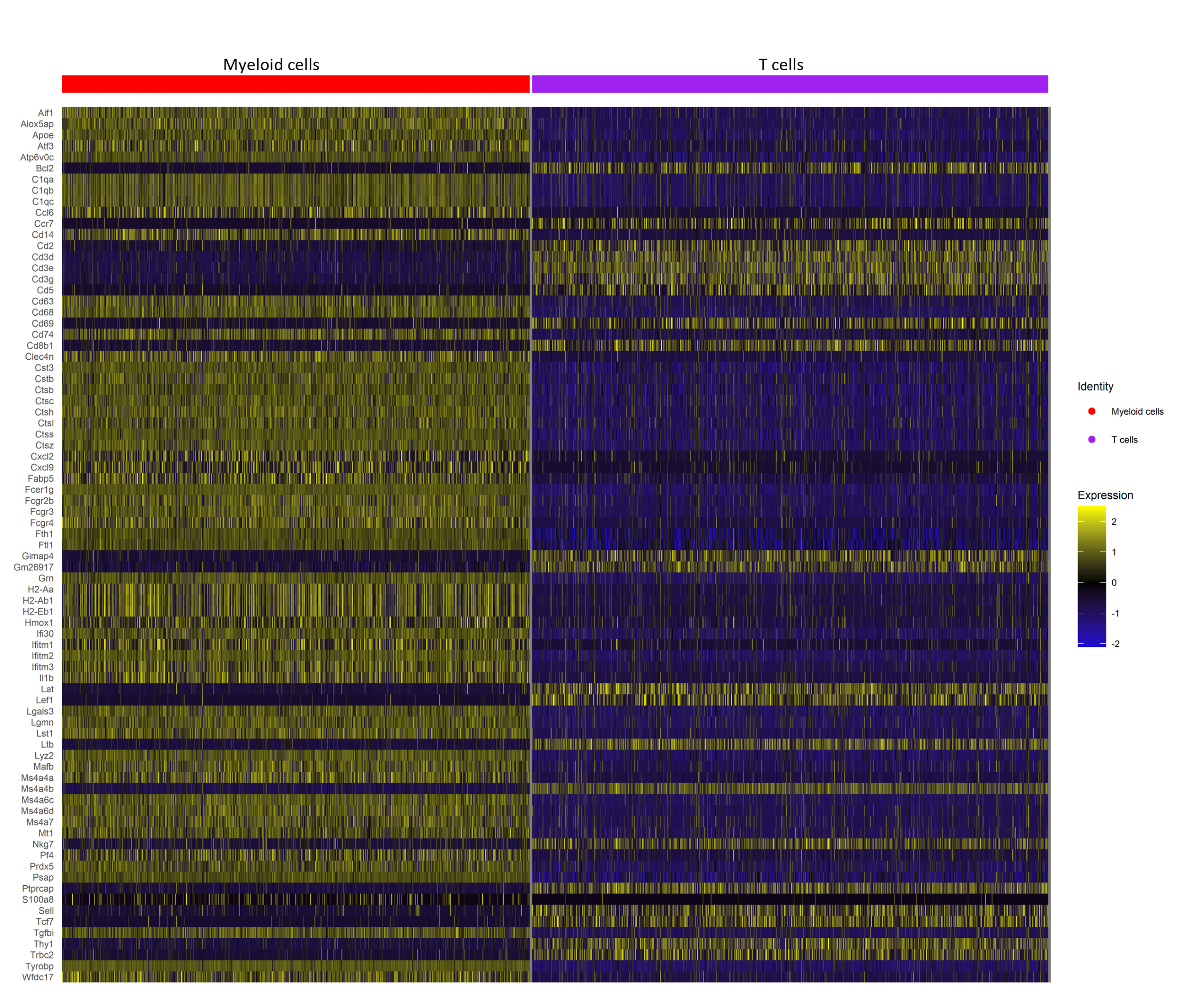
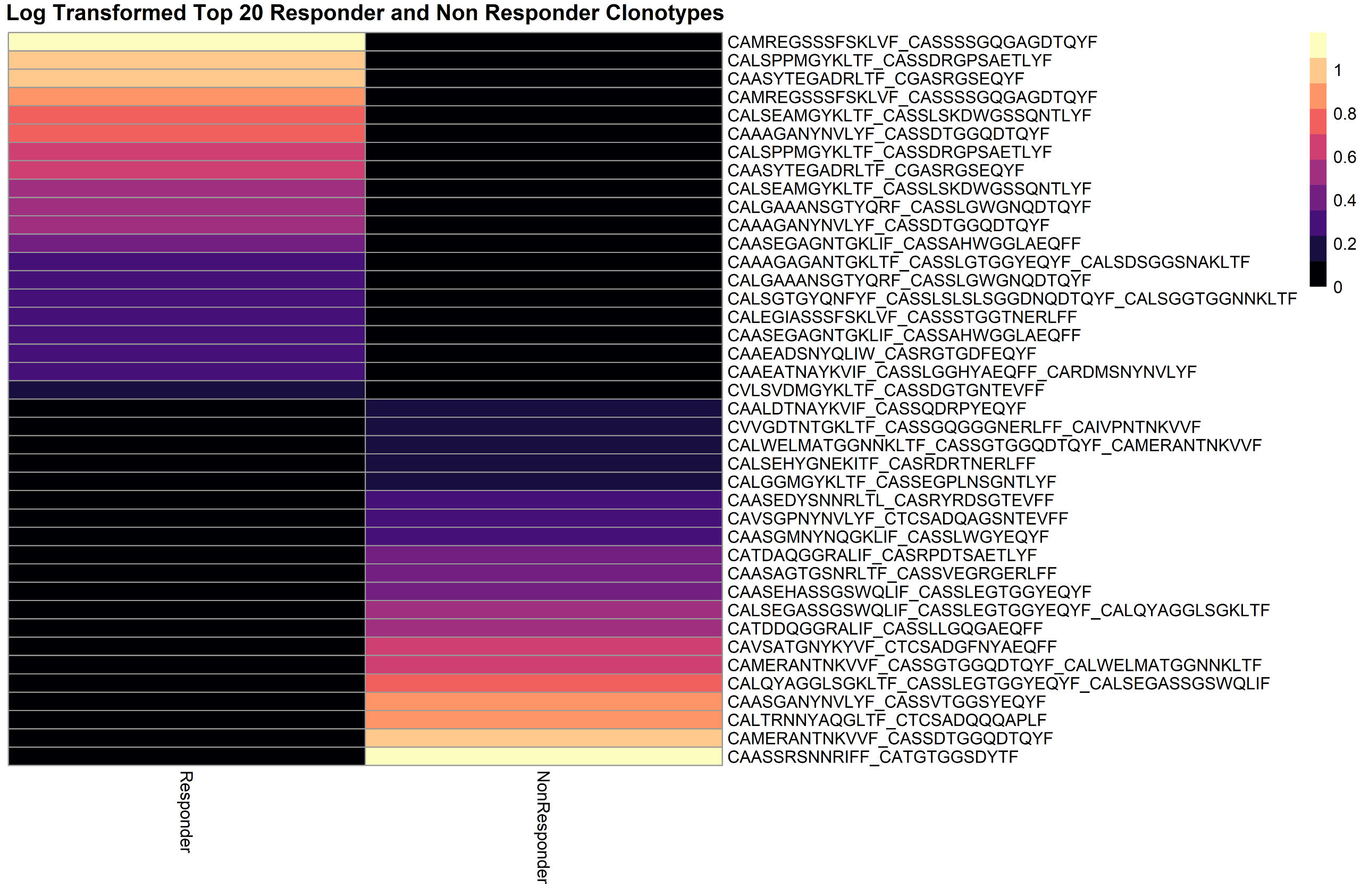
scRNA-seq Analysis
This study explores host intrinsic differences in immune response that influence the effectiveness of anti-PD-L1 treatment in head and neck squamous cell carcinoma (HNSCC). Using a mouse model injected with A223 cells, single-cell RNA sequencing and TCR sequencing were performed on T cells from Responder and Non-Responder mice. Analysis revealed distinct TCR repertoires between responders and non-responders, with expanded clonotypes differing significantly. Gene expression profiling showed that Responder T cells had distinct immunologically relevant upregulated genes, whereas myeloid cell expression profiles were more similar. Contamination issues in T cell sorting were noted, prompting plans for improved methodologies. These findings highlight that intrinsic host factors drive differences in T cell expansion and gene expression, potentially explaining varied responses to immunotherapy.